Automated String Clean Up with rmBadStrings()
PhyloTree_vignette_2.RmdIntroduction
The rmBadStrings functions
(rmBadStrings_1(), rmBadStrings_2(), and
rmBadStrings_3()) can be used to automatically clean a DNA
string set that would otherwise be unsuitable for analysis. These
functions automatically remove strings that are mismatched with other
strings and those whose distances return NaN values or are
considered outliers.
Subset Data
# query the data using the taxon name
specdata <- querySpecData("Nepenthes")
# subset results that only have nucleotides from the matK region
specdata <- subset(specdata, markercode == "matK")
# get one observation per species
specdata <- getSpeciesRepr(specdata)
Manipulate DNA Strings
# generate a DNA bin
DNABin <- genDNABin(specdata)
# use the DNA bin to create a DNA string set
DNAStringset <- genDNAStringSet(DNABin)
# automatically manipulate the DNA strings
DNAStringSet_manip <- ManipStringSet(DNAStringset)
String Clean Up
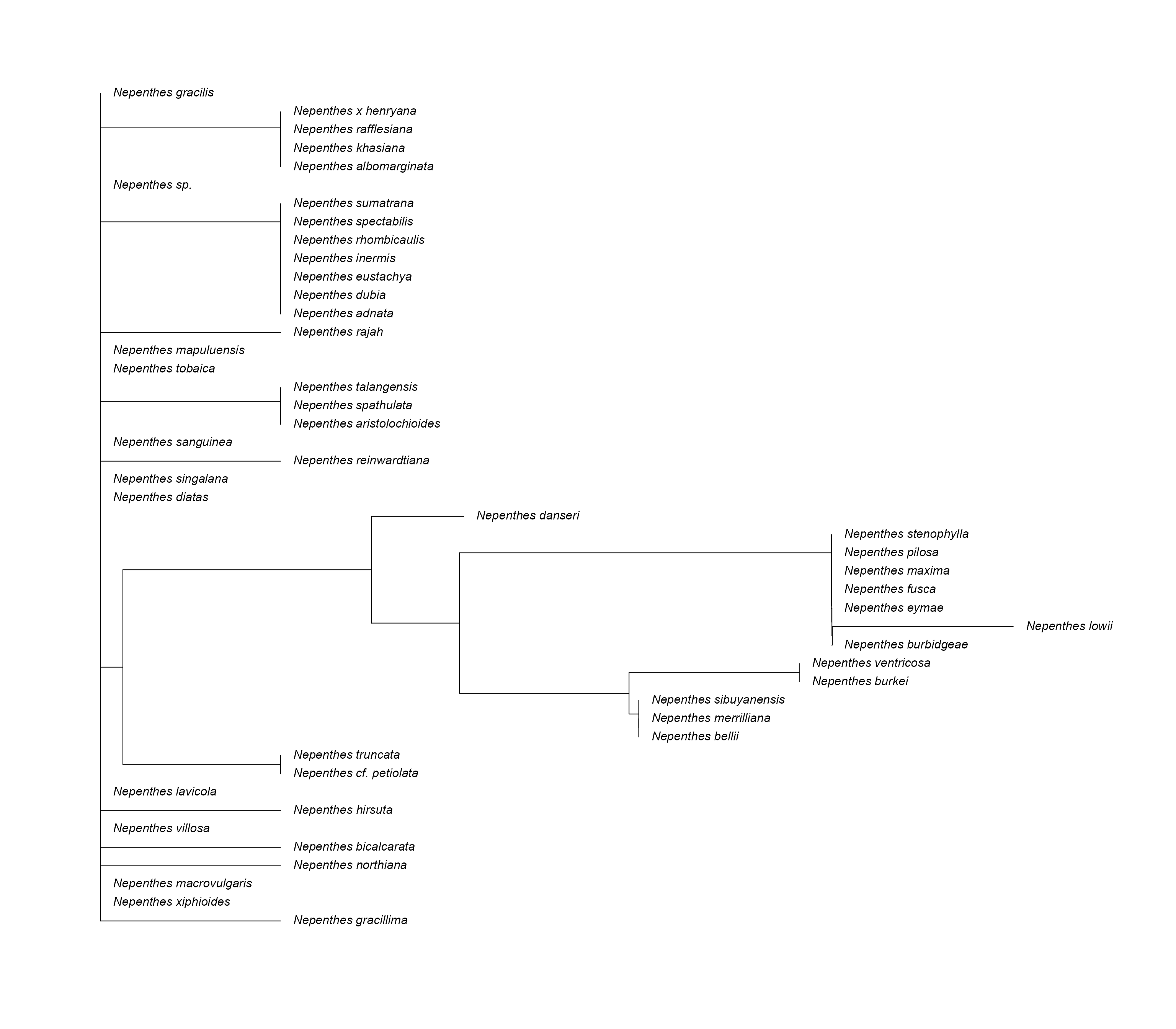
At this point attempting to create a phylo tree will result in an
error. Using DECIPHER::BrowseSeqs(DNAStringSet_manip) to
view the string set will reveal a series of mismatched and fragmented
strings. Using rmBadStrings_3() will automatically remove
all mismatched strings. After which, a tree can be created and and
plotted.
# use the function to remove unsuitable strings and store into an object
BadStringsRemoved <- rmBadStrings_3(
DNAStringSet = DNAStringSet_manip,
specimen_dataframe = specdata,
)Create Phylogenetic Tree
# automatically generate a phylo tree
PhyloTree <- genPhytree(DNAStringSet_new)
# change the label names
PhyloTree$tip.label <- specdata_new$species_name
# plot the phylo tree
plot(
PhyloTree,
label.offset = 0.0001,
cex = 1
)
Remove Outliers
The rmBadStrings functions also have optional arguments
to remove strings whose DNA distances are considered outliers.
rmOutliers is a logical argument that when set to
TRUE will automatically remove outliers as well as
performing its regular processes. max_Z_score is a
numerical value that allows the user to change the maximum Z score for
each string’s DNA distance. The default value for this argument is 3 as
a score higher than this is generally considered an outlier.
Remove Outliers (Below 3 Z-Score)
# use the function to remove unsuitable strings and remove outliers
BadStringsRemoved <- rmBadStrings_3(
DNAStringSet = DNAStringSet_manip,
specimen_dataframe = specdata,
rmOutliers = T
)
#> [1] "Outlier strings detected and removed: 68"Create Phylogenetic Tree
# automatically generate a phylo tree
PhyloTree <- genPhytree(DNAStringSet_new)
# change the label names
PhyloTree$tip.label <- specdata_new$species_name
# plot the phylo tree
plot(
PhyloTree,
label.offset = 0.0001,
cex = 1
)
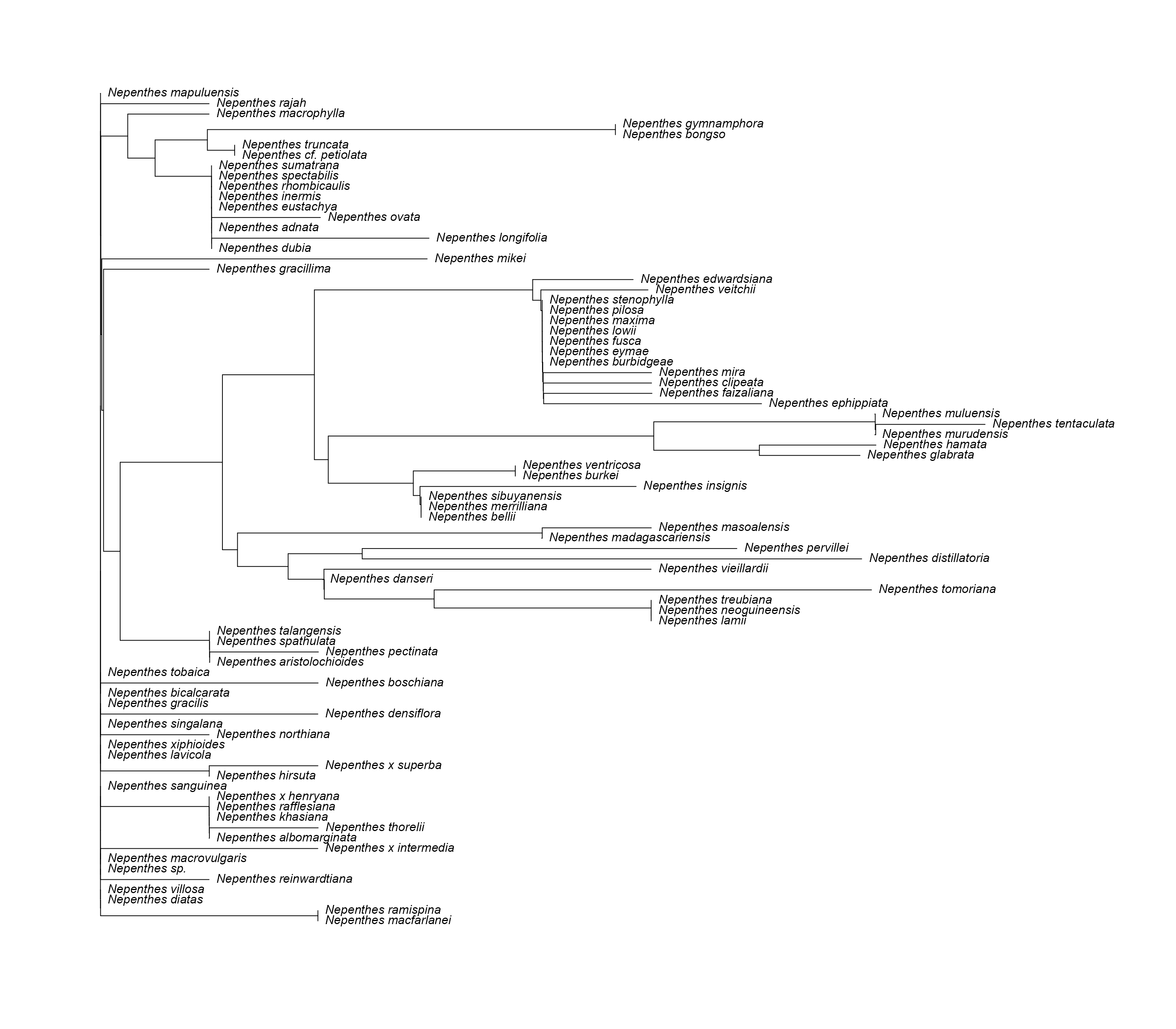
Remove Outliers (Below 2 Z-Score)
# use the function to remove unsuitable strings and remove outliers
BadStringsRemoved <- rmBadStrings_3(
DNAStringSet = DNAStringSet_manip,
specimen_dataframe = specdata,
rmOutliers = T,
max_Z_score = 2
)
#> [1] "Outlier strings detected and removed: 6"
#> [2] "Outlier strings detected and removed: 15"
#> [3] "Outlier strings detected and removed: 18"
#> [4] "Outlier strings detected and removed: 23"
#> [5] "Outlier strings detected and removed: 26"
#> [6] "Outlier strings detected and removed: 27"
#> [7] "Outlier strings detected and removed: 46"
#> [8] "Outlier strings detected and removed: 47"
#> [9] "Outlier strings detected and removed: 52"
#> [10] "Outlier strings detected and removed: 68"
#> [11] "Outlier strings detected and removed: 71"
#> [1] "Outlier strings detected and removed: 10"
#> [2] "Outlier strings detected and removed: 26"
#> [3] "Outlier strings detected and removed: 28"
#> [4] "Outlier strings detected and removed: 35"
#> [5] "Outlier strings detected and removed: 38"
#> [6] "Outlier strings detected and removed: 40"
#> [7] "Outlier strings detected and removed: 61"
#> [8] "Outlier strings detected and removed: 65"
#> [1] "Outlier strings detected and removed: 30"
#> [1] "Outlier strings detected and removed: 6"
#> [2] "Outlier strings detected and removed: 11"
#> [3] "Outlier strings detected and removed: 14"
#> [4] "Outlier strings detected and removed: 17"
#> [5] "Outlier strings detected and removed: 23"
#> [6] "Outlier strings detected and removed: 27"
#> [7] "Outlier strings detected and removed: 28"
#> [8] "Outlier strings detected and removed: 33"
#> [9] "Outlier strings detected and removed: 35"
#> [10] "Outlier strings detected and removed: 36"
#> [11] "Outlier strings detected and removed: 40"
#> [12] "Outlier strings detected and removed: 52"
#> [13] "Outlier strings detected and removed: 55"
#> [14] "Outlier strings detected and removed: 59"
#> [15] "Outlier strings detected and removed: 60"Create Phylogenetic Tree
# automatically generate a phylo tree
PhyloTree <- genPhytree(DNAStringSet_new)
# change the label names
PhyloTree$tip.label <- specdata_new$species_name
# plot the phylo tree
plot(
PhyloTree,
label.offset = 0.0001,
cex = 1
)