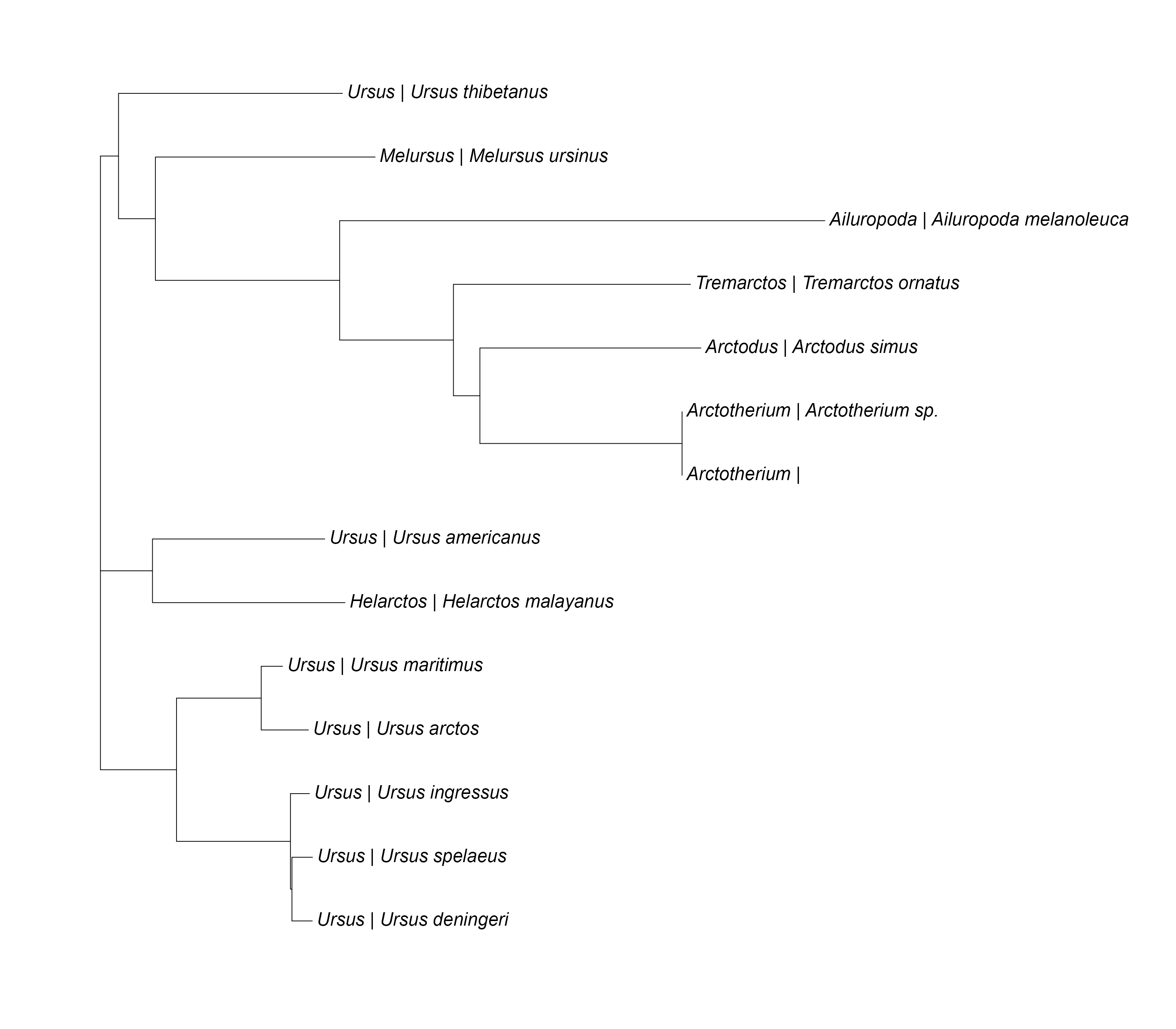
Quick Tree Building
PhyloTree_vignette_1.RmdIntroduction
The PhyInsight package is used to efficiently conduct
DNA sequence analyses. In this vignette, use of the package functions in
order to create a phylogenetic tree will be demonstrated. The functions
in this package are used to manipulate DNA strings, remove mismatched
strings, and remove strings that cause distance outliers where
necessary. These processes are performed automatically when the
appropriate functions are used and can save the user much time and
effort.
Any taxon can be used provided the DNA strings are in the correct
format. The Ursidae family will be used as an example of how one can use
PhyInsight to manipulate DNA strings and build a
phylogenetic tree in just four functions once the specimen dataframe is
ready.
Subset Data
# query the data using the taxon name
specdata <- querySpecData("Ursidae")
# check the number of DNA marker codes
sort(table(specdata$markercode))
#>
#> 16S D-loop ND1 ND2 ND3 ND4 ND4L ND5-0 ND6 atp6 COII COXIII CYTB COI-5P
#> 4 9 14 14 14 14 14 14 14 39 41 41 42 523
Choosing which gene region(s) to use is an important step and will
vary depending on the research question and the taxa. In this example,
only sequences from the COI-5P region will be used for the
analysis. Also, getSpeciesRepr() is used to automatically
select representative observations for each species.
# subset results that only have nucleotides from the COI-5P region
specdata <- subset(specdata, markercode == "COI-5P")
# get one observation per species
specdata <- getSpeciesRepr(specdata)
Manipulate DNA Strings
Here, functions from the package are used to automatically create a DNA bin, create a DNA string set object, then automatically manipulate the strings.
# generate a DNA bin
DNABin <- genDNABin(specdata)
# use the DNA bin to create a DNA string set
DNAStringset <- genDNAStringSet(DNABin)
# automatically manipulate the DNA strings
DNAStringSet <- ManipStringSet(DNAStringset)
Create Phylogenetic Tree
Here, the genPhytree() function is used to automatically
create a phylo tree object. The labels names, offset, and size can also
be edited.
# automatically generate a phylo tree
PhyloTree <- genPhytree(DNAStringSet)
# change the label names
PhyloTree$tip.label <- paste(specdata$genus_name, "|", specdata$species_name)